
Last updated on December 11th, 2022 at 05:53 pm
Are you wondering What are difference between electric and hybrid cars? Well, in this article, we are discussing the difference between electric and hybrid vehicles, What’s better, hybrid or electric? That is; the hybrid vs electric cars pros and cons and, lastly, the difference between hybrid and plug in hybrid. I will share a comprehensive hybrid electric vehicle pdf that you might find very useful.
What are Difference Between Electric and Hybrid Cars
When you tend to shop for eco-friendly vehicles, you will likely have questions about electric and hybrid cars. Although electric cars have been a buzzword for some time now, you might have heard of hybrid cars available in your locality for a longer time.
However, as regards some confines on modern-day battery technology, electric cars were, for a longer time, incapable of offering the sort of usage range required by an average user. But, with today’s technological advancements, electric vehicles have been in the process of offering drive ranges that make them highly attractive for the average individual.
Similarly, hybrid vehicles offer what is known to be the middle-ground solution to individuals that suffer from range anxiety, as well as the fear that they might get stranded on the road without a charging solution. Additionally, hybrid vehicles use electric efficiency and fuels as substitute energy sources.
Notwithstanding, this informative guide would tend to examine and proffer solutions to these typical questions and issues such as; what’s the difference between hybrid and electric cars, what’s better, hybrid or electric, what are the advantages of a hybrid car, the difference between hybrid and plug-in hybrid, hybrid vs. electric cars pros and cons, hybrid electric vehicle pdf, and more.
Without much ado, let’s get started!
Related Articles: Toyota solid State Battery: What Happened to Toyota’s solid-state battery?

What’s The Difference Between Hybrid And Electric Cars?
It is without a doubt that the difference between hybrid and electric vehicles is clear.
Invariably, EV models run on one battery power source, whereas a hybrid utilizes ICE and battery to maintain fuel efficiency.
The incentives received by EV owners are the highest in the automobile world as you need to charge it, whereas models of hybrids passively regenerate their batteries.
For smooth and faster acceleration, the EV is the answer, but a hybrid would do the same to an extent because of poor handling and lack of power.
The EV has zero emission, unlike the hybrids that combine ICE and battery systems and, as such, produce emissions that affect the ecological system.
Hopefully, the above is helpful!
What’s Better, Hybrid Or Electric?
EVs have a null tendency to emission whereas the hybrids have high emission, but as regards the better, it is subject to your choice, financial status, and lifestyle. Perchance, a hybrid is suitable if you want better fuel efficiency but don’t prefer to charge your vehicle. Similarly, if you drive hundreds of miles daily, EV is preferable. In terms of cost, a hybrid is less costly than an EV if you want to purchase a car. So far, each has the edge over the other, and the two vehicles are good.
Consider all available options, pros, and cons of these vehicles as regards your income and lifestyle before arriving at a conclusion and decision, in any case!
What Are The Advantages Of A Hybrid Car
Hybrid cars have become so common even to the average individual since their manufacturing cost has declined. Here are the most exciting advantages of having a hybrid vehicle!
1) Financial Benefits
Having a hybrid car would make you pay lower tax bills and some exceptions from congestion charges that might emanate from a small amount of money spent on gasoline. Similarly, hybrid cars are inexpensive because they have incentives and credits, unlike other standard vehicles.
2) Less Maintenance And Cost Efficiency
Using a high voltage battery pack and an ICE to propel the hybrid vehicle makes it have less wear and tear on its components, unlike the standard or the classic car. With a hybrid vehicle, you would incur fewer repairs and fewer regular maintenance costs.
3) Built From Very Light Materials
A hybrid vehicle is designed with light materials, requiring less energy. The hybrid vehicle’s engine is more delicate and smaller than the traditional vehicle.
4) Less Dependence On Gasoline
Since a hybrid car is cleaner as it requires less gasoline to run, thus, fewer emissions are produced and, in like manner, less gasoline dependence. Remember that hybrid car uses dual-power or twin-power, so an alternative is a very high advantage to fossil fuel consumption.
5) Friendly To The Environment
A hybrid vehicle runs on a double or twin-power engine (electric motor and gasoline engine) that reduces fuel consumption to a minimal level and conserves energy. This advantage is one of the finest over traditional vehicles (gasoline-powered vehicles). In addition, it has a more improved mileage and runs cleaner, making it friendly to the environment.
6) Higher Resale Value
As gasoline prices rise daily, it is evident that many people have turned towards hybrid cars as effective options. As regards that, hybrid cars have begun to command higher resale values in the automobile world. Perchance you still have a traditional vehicle; it is time you switch to a better hybrid car.
7) Electric-Only Drive
The hybrid vehicle can also use electricity entirely as it begins at a low speed. However, it is more efficient when operating at higher rates during internal combustion. So, the efficiency of your hybrid vehicle works on the electric-only drive system.
8) Range Anxiety Not An Issue
Recently, range anxiety is no longer a common issue among PHEV or hybrid vehicle owners or drivers. In essence, when your battery runs low on charge in your hybrid vehicle, then the car would rely on the alternative source of the power of ICE. This quick switch in control is comfortable where gas or electric charging stations are not readily accessible.
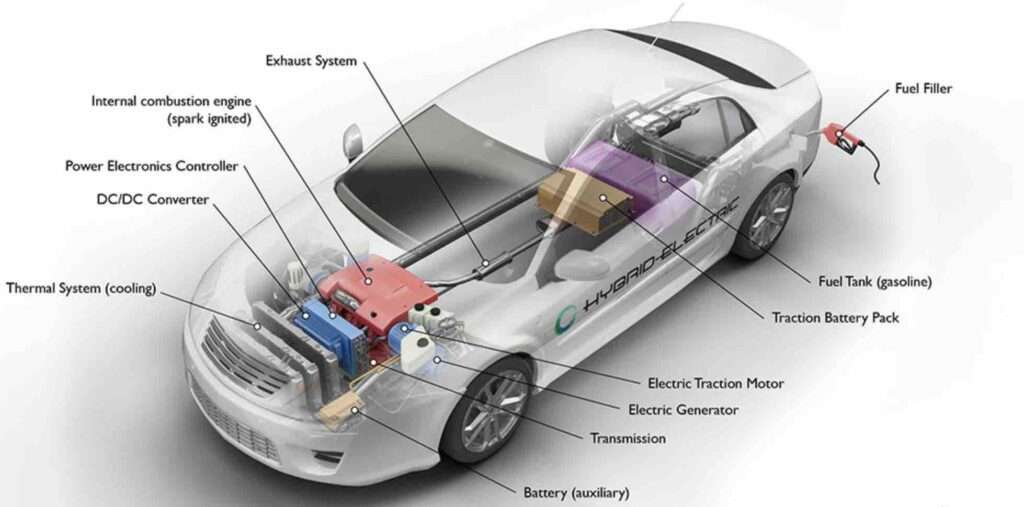
What Are The Disadvantages Of A Hybrid Car
Be it as it may be, hybrid cars have disadvantages popular to the standard regular or traditional vehicles regardless of the type of hybrid fuel each vehicle uses.
Here you have their disadvantages;
1. Hydrogen Fuel Cell Problems
The source of hydrogen is both clean and dirty sources such as solar and wind and coal and natural gas, respectively. However, sourcing from coal and natural gas can destabilize the ecological motive for using hydrogen fuel cell cars. Notwithstanding, the production of hydrogen cells and hydrogen stations is expensive.
2. Expensive To Buy
Without much ado, hybrid cars are more expensive than standard or traditional vehicles. They cost about $5000 to $10000 more than regular vehicles; this offsets lower running costs and some tax exceptions.
3. Less Power
A hybrid vehicle is a dual-powered engine, has its primary power source being a gasoline engine; when compared to a standard car (single-engine), it is less in power. Similarly, the electric motor, of course, has less energy than a fully electric vehicle. Hence, it is not technically meant or suited for speed and acceleration, but for city driving, in any case.
4. Pricey Battery Replacement
The batteries of hybrid vehicles are rare to buy, according to Green Car Report. So, if you want to replace yours, it could be pricey.
5. High Cost Of Maintenance
The presence of twin power and technological advancement makes it difficult for certified mechanics to repair such cars. On the other hand, the maintenance cost is much higher since finding a mechanic with such skills is very frustrating.
6. Accidents From High Voltage Batteries
The high voltage battery inside your hybrid vehicle could be dangerous to you, the driver, in an accident. There are high tendencies that you could get electrocuted as it is complex to rescue passengers or drivers from such an event.
Difference Between Hybrid And Plug-In Hybrid
The significant difference between plug-in-hybrid and hybrid vehicles is mainly the cost, battery size, and, perchance, their electric batteries.
Let’s dive into the difference between hybrid and plug-in-hybrid without much ado!
- The purpose of battery – In plug-in-hybrid, the principal source of power is the electric battery. So, if your plug-in-hybrid runs down, the ICE instantly takes control. On the other hand, electric power needful for lower speeds in the full hybrid vehicle is less.
- The battery cost and size – The electric battery in hybrid is smaller and less expensive than the plug-in-hybrid electric battery. However, the plug-in hybrid car is more expensive than a full hybrid.
- The recharging capabilities of the batteries – The hybrid car would charge its battery through a regenerative braking system and, as such, take the heat produced by the braking event and convert it to stored electricity in its battery. Similarly, the plug-in hybrid would acquire a little charge through the regenerative braking system. However, it relies on more external power since it is a larger battery.
Note: The similarity between the plug-ins and the full hybrids is that both vehicles become gasoline-powered when their batteries run down.
Hybrid Vs. Electric Cars Pros And Cons
Let’s quickly get started as I consider some factors that would help you decide which is preferred for you, hybrid vs. electric cars:
· Costs Difference
Since the hybrid have both electric and internal combustion engine (ICE) to maintain, its costs are high in maintenance aspect but less upfront. Electric vehicles can be more expensive but have a few moving parts that might break down over time. As much as the EVs have features, labor costs are high.
· Availability Of Charging Stations
Consider the electric car if you intend to go on a road trip. It is ideal for making sure there are charging stations along that route. However, if there are none, then the hybrid is the answer and a better option for you to avoid frustrating situations.
· Home Owner Vs. Renter
Considering that you own your home, nothing stops you from installing a charger on your EV or PHEV overnight. But, this would be different if you don’t own your home or are on rent; perchance chargers are available in your community; if not, you should opt for a hybrid car.
· Emission Level
Electric vehicles have significantly lower emission rates compared to hybrid and ICE vehicles. So far, hybrid cars have a higher tendency to affect the ecological level due to hydrogen production from dirty sources like coal and natural gas.
Concluding on What are Difference Between Electric and Hybrid Cars
In my final words, a hybrid car runs on an electric motor and ICE with separate batteries for each system. Perchance, an electric vehicle has only a battery as the power source as it utilizes an electric motor to run.
Invariably, electric and hybrid vehicles have a lower environmental impact than standard or regular cars (ICEs). Notwithstanding, the EV and the hybrid have distinctive features in upfront costs, charging/refueling costs, driving range, and maintenance costs.
So far, if you intend to buy any of these vehicles, consider factors such as your environment or locality, choice, pocket, and lifestyle and study through the above work to be precise on the right car for you. Always make the right choice to complement your life; enjoy your ride, in any case! Do like and share this post with others!
For more on this topic, check out this hybrid electric vehicle pdf. I think you will love it.

Uchenna is a Radiographer and Auto parts mechanic who recently got his automotive diploma as an auto repair technician, and since then, has worked on fixing various car problems.
Working as just a radiographer, Uchenna didn’t just get all the fulfillment he desired, because he truly loved doing things tilted toward cars. As a kid, he would take apart his toy cars to see how they worked and would spend hours tinkering with his bike.
So, in 2017 he made the tough decision to become an auto mechanic. He threw himself into his studies and now loves every aspect of what he does.
He gets to work with his hands, solving problems and bringing cars back to life, and sharing his knowledge and easy quick-fix guide online are all part of what makes him feel fulfilled.


